Tutorial
Build example
Running ergo with inline molecule
Write matrices to .mtx files
Get information about HOMO/LUMO orbitals in Gabedit format
Using a previously computed density as a starting guess for a new calculation
Energy surface for H2
Extract source code from tar file
tar -xzf ergo-3.3.1.tar.gz
Build the executable. If you do not have lapack installed you can use the internal
library by configuring with the flag
"--enable-linalgebra-templates". Note that the internal library is
not very well optimized, so if better performance is needed you
should link to some optimized BLAS/LAPACK library.
cd ergo-3.3.1
./configure --enable-linalgebra-templates && make
You may want to check that the build was ok:
make check
This may take a while. Both unit tests and tests running complete
SCF calculations will be performed when running "make check".
Let us create a new directory in which we will run ergo.
cd ..
mkdir rundir
cd rundir
We need the executable
cp ../ergo-3.3.1/source/ergo .
Let us create a file "run_ergo.sh" with the following content:
#!/bin/sh
./ergo <<EOINPUT
molecule_inline
O 0.0 0.0 0.0
H -1.809 0.0 0.0
H 0.453549 1.751221 0.0
EOF
basis = "6-31Gss"
run "HF"
EOINPUT
Here, the lines between "molecule_inline" and "EOF" specifies the
geometry of the molecule; in this case a water molecule. Then we
need to specify which basis set we want to use; in this case the
"6-31G**" basis set. Finally, the line 'run "HF"' will start a
Hartree-Fock calculation with the molecule and basis set specified
above.
Do not forget to set executable permissions to the script file:
chmod u+x run_ergo.sh
We are now ready to run the calculation.
./run_ergo.sh
This will generate the following files:
ergoscf.out
density.bin
"ergoscf.out" is the output file where information of the run is written. It should contain the lines
RESC CONVERGED after 12 iterations.
RESC FINAL ENERGY: -76.02264311439
Then we know that the calculation has converged.
In the file "density.bin", electron density and basis set
information is stored so that a new calculation can
use it as starting guess (using the
"initial_density" parameter).
Now we want to output matrices in MatrixMarket (.mtx) format. We
would like to have Fock, density, and overlap matrices. If you do not
remember the name of the input parameters run:
./ergo -h
From here you find the variables "create_mtx_files_D",
"create_mtx_files_F", and "create_mtx_file_S" under the list
"scf".
To get information about any input parameter run:
./ergo -d list.name_of_variable_in_list
To output matrices, revise your run_ergo.sh script file as follows:
#!/bin/sh
./ergo <<EOINPUT
molecule_inline
O 0.0 0.0 0.0
H -1.809 0.0 0.0
H 0.453549 1.751221 0.0
EOF
basis = "6-31Gss"
scf.create_mtx_files_D = 1
scf.create_mtx_files_F = 1
scf.create_mtx_file_S = 1
run "HF"
EOINPUT
We are now ready to run the calculation.
./run_ergo.sh
Note that the new output will be appended to the ergoscf.out file if
it already exists. The following .mtx files will be generated:
S_matrix.mtx
F_matrix_X.mtx, X = 1, 2, ...
D_matrix_X.mtx, X = 1, 2, ...
The S_matrix.mtx file contains the overlap matrix in mtx format.
The files F_matrix_X.mtx, X = 1,2, ... contain the Fock matrix for each SCF
iteration as specified by the serial number X. The
file with the highest number contains the matrix
for the last SCF iteration.
The files D_matrix_X.mtx, X = 1,2, ... contain the density matrix for each SCF
iteration as specified by the serial number X. The
file with the highest number contains the matrix
for the last SCF iteration.
Ergo can produce output in a format compatible with the Gabedit program (see
http://gabedit.sourceforge.net/). To generate information about HOMO/LUMO orbitals in Gabedit format, the following input file could be used:
#!/bin/sh
./ergo <<EOINPUT
molecule_inline
O 0.0 0.0 0.0
H -1.809 0.0 0.0
H 0.453549 1.751221 0.0
EOF
basis = "6-31Gss"
scf.output_homo_and_lumo_eigenvectors = 1
run "HF"
EOINPUT
Now run the program:
./run_ergo.sh
This will generate a file called "gabeditfile.gab" which can be used in the Gabedit program.
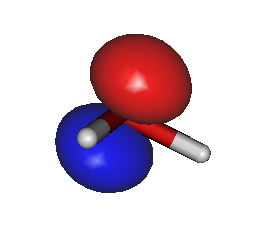
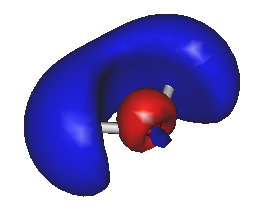
For example, to generate HOMO/LUMO images, run Gabedit and select the "Display Geometry/Orbitals/Density/Vibration" option, then select "Orbitals --> Read geometry and orbitals from a Gabedit file", and choose your new file "gabeditfile.gab" that was created by Ergo. Then choose reasonable values for the "Box & Grid" and "Iso-Value" parameters in the Gabedit dialog boxes. The resulting images can look like this:
During each Ergo calculation, the current electron density is saved in a file named "density.bin". This file can later be used as starting guess for a new calculation. To use a previously computed density as a starting guess for a new calculation, use the option "initial_density", for example like this:
#!/bin/sh
./ergo <<EOINPUT
molecule_inline
O 0.0 0.0 0.0
H -1.809 0.0 0.0
H 0.453549 1.751221 0.0
EOF
initial_density = "density.bin"
basis = "6-31Gss"
scf.output_mulliken_pop = 1
run "HF"
EOINPUT
If a good starting guess is used, fewer SCF cycles are typically needed to reach convergence.
We wish to see how the energy depends on internuclear distance for H
2.
We are performing calculations for two H atoms with distance 2d between them. Example with d=2:
molecule_inline
H 2 0.0 0.0
H -2 0.0 0.0
Restricted calculations
When the number of electrons is even, like in our H
2 case where we have two electrons, Ergo performs a spin-restricted calculation by default. Therefore, we do not need to specify any input parameter to get a restricted calculation.
We will set the convergence threshold for the SCF procedure:
scf.convergence_threshold = 1e-5
Final code:
./ergo << EOINPUT
molecule_inline
H 2 0.0 0.0
H -2 0.0 0.0
EOF
basis = "6-31Gss"
scf.convergence_threshold = 1e-5
run "HF"
EOINPUT
When the calculation has finished you can find the final energy in the output file ergoscf.out.
In restricted calculations alpha and beta electrons are forced to occupy the same spatial molecular orbital. As a result the energy is too high for well separated hydrogen atoms.
Unrestricted calculations
Even though the numbers of alpha and beta electrons are equal, we should in this case use unrestricted SCF calculations:
scf.force_unrestricted = 1
By default Ergo uses the same initial density matrix guess for alpha and beta electrons. To randomly perturb the initial guess use the following option:
scf.starting_guess_disturbance = 0.01
We will set this parameter for the computation with maximum internuclear distance (in our example the maximum internuclear distance is 8):
./ergo << EOINPUT
molecule_inline
H 4 0.0 0.0
H -4 0.0 0.0
EOF
basis = "6-31Gss"
scf.convergence_threshold = 1e-5
scf.force_unrestricted = 1
scf.starting_guess_disturbance = 0.01
run "HF"
EOINPUT
For computations with lower internuclear distances we will use as initial guesses density matrices from previous SCF calculations (with larger internuclear distances):
./ergo << EOINPUT
molecule_inline
H 2 0.0 0.0
H -2 0.0 0.0
EOF
basis = "6-31Gss"
scf.convergence_threshold = 1e-5
scf.force_unrestricted = 1
initial_density = "density.bin"
run "HF"
EOINPUT
One hydrogen atom
To find the energy for a single hydrogen atom, we should also specify the "spin_polarization", which is defined as the difference between the number of alpha and beta spin electrons. (For equal number of alpha and beta electrons, spin_polarization = 0).
Therefore add in the script:
spin_polarization = 1
Final code:
./ergo << EOINPUT
molecule_inline
H 0.0 0.0 0.0
EOF
basis = "6-31Gss"
scf.force_unrestricted=1
spin_polarization=1
run "HF"
EOINPUT
Energy surface
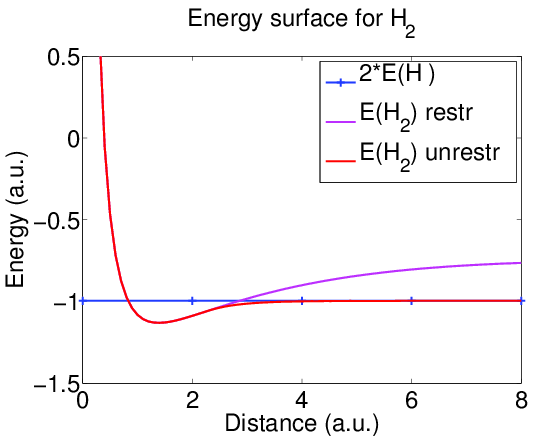
The minimum of the energy occurs at internuclear distance near 1.4 a.u., which gives the equilibrium geometry.
The Ergo tutorial page ends here. If you have any questions, suggestions, or other comments regarding this tutorial, please contact us (see contact page).
We hope you will enjoy using Ergo!